















Nombre: Dermatomiositis
Tipo: Enfermedades del tejido conectivo
Categoría:
Imagen:

Gráfico:



Información: Dermatomiositis
La dermatomiositis es una enfermedad inflamatoria, de probable base autoinmune, que se caracteriza por la afectación muscular con afectación predominante de las cinturas musculares pelvianas y escapulares, por el desarrollo de lesiones cutáneas características y por la posible afectación de órganos internos. El término de polimiositis se utiliza en aquellos pacientes en los que no existe afectación cutánea.
La polimiositis y dermatomiositis son clasificadas como una misma enfermedad con un espectro clínico diferente, clasificándose en 6 tipos según las manifestaciones y caraterísticas clínicas (tabla VI).
La dermatomiositis es relativamente infrecuente, con una incidencia anual de 5-10 casos por millón y año y una prevalencia de 5-8 casos por cada 100.000 habitantes, es más frecuente en mujeres y se puede manifestar en cualquier edad, existiendo dos picos de mayor incidencia uno en la infancia (<10 años) y otro en la edad adulta (45-60 años). El diagnóstico de la dermatomiositis debe establecerse según unos criterios diagnósticos que son útiles para la realización de estudios y que están resumidos en la tabla VII.
Tabla VI
Clasificación de la dermatomiositis/polimiositis
I
Polimiositis idiopática primaria
II
Dermatomiositis idiopática primaria
III
Polimiositis o dermatomiositis asociada a neoplasia
IV
Dermatomiositis o polimiositisjuvenil
V
Polimiositis o dermatomiositis asociada a otra enfermedad de tejido conectivo (Síndrome de solapamiento)
VI
Dermatomiositis amiopática
Tabla VIII Manifestaciones clínicas de la dermatomiositis/polimiositis
Cutáneas
Poiquiloderma
Pápulas de Gottron
Rash heliotropo
Engrosamiento de la cutícula
Manos de mecánico
Fenómeno de Raynaud
Músculo-esqueleticas
Artralgias y artritis
Debilidad muscular de cintura escapular y pelviana
Gastrointestinales
Disfagia con regurgitación nasal
Perforación del tracto gastrointestinal secundaria a vasculitis (dermatomiositis juvenil)
Pulmonares
Reducción de la capacidad ventilatoria secundaria a la afectación de músculo diafragmático y músculos accesorios
Neumonía por aspiración
Infecciones secundarias a la terapia
Fibrosis intersticial
Cardíacas
Miocarditis sintomática infrecuente asociada con arritmias y insuficiencia cardíaca congestiva
Cor pulmonale secundario a enfermedad pulmonar intersticial
Tabla VII
Criterios diagnósticos de DMS de Bohan y Peter
Debilidad simétrica proximal, progresando durante semanas o meses
Biopsia muscular que evidencia una miopatia inflamatoria
Aumento de los niveles séricos de enzimas musculares
Hallazgos electromiográficos de miopatia
Erupción cutánea típica de dermatomiositis
PM/DM definitiva: 4 criterios
PM/DM probable: 3 criterios
PM/DM posible: 2 criterios
Criterio 5 necesario para considerar DM
Manifestaciones clínicas: La dermatomiositis/polimiositis tiene una expresión clínica variada que está resumida en la tabla VIII.
Manifestaciones cutáneas
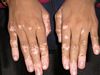
Pápulas de Gottron: Se observan en más del 80% de pacientes con dermatomiositis y se consideran patognomónicas de la enfermedad. Consisten en pápulas y placas violáceas localizadas en la cara dorsal de las articulaciones interfalángicas y metacarpofalángicas. Se considera el signo de Gottron la presencia de un eritema macular violáceo en la misma localización y que también puede verse localizado alrededor de otras articulaciones incluyendo codos, rodillas, tobillos.
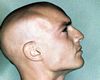
Rash heliotropo :Se observa hasta en el 60% de los pacientes consiste en el desarrollo de áreas de poiquilodermia afectando a párpados y región periorbitaria, con marcada fotosensibilidad, ocasionalmente se asocia con la presencia de marcado edema, especialmente cuando se observa en una dermatomiositis asociada a neoplasia.
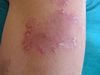
Poiquilodermia:Constituye un hallazgo clínico cutáneo muy característico de la dermatomiositis. Consiste en el desarrollo de placas cutáneas con áreas hiper e hipopigmentadas, atroficas y con telangiectasias prominentes, con marcada fotodistribución, afectando a la región de la "V" del escote (signo del chal) y base del cuello.
Afectación de la cutícula : La cutícula periungueal se puede observar engrosada con presencia de telangiectasias que a simple vista o mediante capilaroscopia mediante oftalmoscopio puede demostrar la presencia de pequeños vasos capilares de trayecto tortuoso. También pueden observarse áreas de hemorragia en astilla e infarto.
Manos de Mecánico : consiste en el desarrollo de áreas de hiperqueratosis y fisuración de las caras laterales y palmares de los dedos de las manos, lo cual produce un aspecto de manos sucias similares a las observadas en los mecánicos.
Calcinosis cutis: constituye una complicación tardía que se observa en el 15% de las dermatomiositis de los adultos y en el 60% de dermatomiositis juvenil. Consiste en el deposito de calcio en el tejido subcutáneo y fascia y puede dar lugar al desarrollo de ulceras cutáneas.
Otros hallazgos: Los pacientes con dermatomiositis pueden desarrollar varios tipos de lesiones cutáneas características de la enfermedad pero no patognomónicas que incluyen la presencia de pápulas hiperqueratósicas foliculares de distribución lineal afectando al dorso de manos, vasculitis y fenómeno de Raynaud.
Histología de las lesiones cutáneas: El estudio histológico de las lesiones cutáneas de la dematomiositis muestra cambios similares a los observados en en lupus eritematoso, en las pápulas de Gottron existe una acantosis epidérmica y engrosamiento marcado de la membrana basal que adquiere un aspecto tortuoso con marcada degeneración vacuolar de la membrana basal que en ocasiones puede acompañarse de intensos depósitos de fibrina en la unión dermo-epidérmica. En dermis existe un infiltrado inflamatorio intersticial de predominio linfocítico acompañado de depósitos de mucina y ocasionalmente pueden observarse focos de calcificación.
Afectación sistémica
Afectación muscular: El desarrollo de alteraciones clínicas y analíticas de afectación muscular es un hallazgo frecuente en pacientes con dermatomiositis. Las lesiones cutáneas preceden a la afectación muscular en la mayoría de pacientes con dermatomiositis. La afectación muscular afecta principalmente a los músculos de la cintura escapular y pelviana y generalmente tiene una distribución simétrica. Los pacientes suelen referir debilidad y fatiga así como dificultad o incapacidad para realizar esfuerzos a los que normalmente estaban habituados tales como subir escaleras, levantar los brazos para peinarse o afeitarse, levantarse de una silla, etc. La afectación muscular puede ser progresiva con desarrollo de dificultad en deglución y afectación de los músculos respiratorios. La afectación clínica muscular se acompaña de alteraciones analíticas, electromiográficas y patológicas en las biopsias musculares. Analíticamente se pueden observar elevaciones de enzimas como la CPK, LDH, aldolsa y transaminasas, pero no necesariamente han de estar todas ellas elevadas. La medición de niveles de CPK es probablemente la más útil para determinar la afectación muscular. En pacientes con enzimas normales, la medición de excreción urinaria de creatinina puede reflejar la actividad inflamatoria muscular.
Los estudios electromiograficos son característicos mostrando una innervación nerviosa intacta, lo que es útil para descartar procesos neuropáticos. Las alteraciones electromiográficas características consisten en la presencia de potenciales miopáticos caracterizados por unidades o potenciales de acción polifásicos de baja amplitud o voltaje y de corta duración bajo activación voluntaria, así como un incremento de la actividad espontánea con fibrilaciones, descargas complejas repetitivas y ondas agudas positivas.
La biopsia muscular debe realizarse de músculos clínicamente afectos y va a mostrar un infiltrado inflamatorio endomisial compuesto predominantemente por linfocitos que se distribuyen preferentemente en las áreas perivasculares de los septos interfasciculares. Pueden observarse grados variables de necrosis de fibras en la periferia de los haces musculares.
Otras técnicas exploratorias útiles en la valoración de la afectación muscular incluye la resonancia magnética nuclear y resonancia magnética espectroscópica con fósforo 31, que pueden ser útiles en detectar la afectación muscular y que en el futuro puede que sustituyan a la electromiografía y a la biopsia muscular.
Afectación pulmonar: un 15-30% de pacientes tienen afectación pulmonar en forma de fibrosis intersticial difusa, que se observa especialmente en pacientes con anticuerpos antisintetasa. Los pacientes con dermatomiositis también pueden desarrollar neumonía por aspiración o insuficiencia respiratoria si existe afectación de la musculatura intercostal o diafragmática.
Tabla IX.
Anticuerpos en la Dermatomiositis
Ac Inespecíficos
Ac antinucleares
a títulos bajos, en síndromes de solapamiento se observan a títulos >1:160
Ac específicos
Ac anti-sintetasa (Anti Jo, OJ, PL, KU, KS)
Su presencia se asocia al síndrome antisintetasa
Ac Anti Mi-2 se asocia con el desarrollo de dermatomiositis
Ac Anti 155/140
Asociado a una alta relación con neoplasia
También asociado a dermatomiositis juvenil
Ac Anti PM/SCL
Asociación PM/esclerodermia, esclerodermia solo
Ac asociados a DM
anti-RNP
Enfermedad mixta del tejido conectivo
Anti-Ku
Síndrome de solapamiento PM-SCL, SLE, esclerodermia
Estudios serológicos en pacientes con Dermatomiositis
Los pacientes con dermatomiositis muestran positividad para una gran variedad de anticuerpos (Tabla IX). Los anticuerpos encontrados en estos pacientes pueden clasificarse en los asociados a miositis que también se encuntran en otras enfermedades autoinmunes y los espcíficos de miositis que se encuentran especialmente en este grupo de enfermos. Un 90% de pacientes muestra positividad para los ANA generalmente a títulos bajos. Un 20% muestran anticuerpos anti -Mi-2, dirigido contra un antígeno nuclear de 240-kD, que muestra una alta especificidad par la dermatomiositis (el 95% de pacientes que lo presentan tienen dermatomiositis). Los pacientes con polimiositis y menos frecuentemente los afectos de dermatomiositis presentan anticuerpos contra diferentes tipos de aminoacil-tRNA sintetasas, que constituyen un grupo de enzimas que unen la tRNA a el aminoácido correspondiente (catalizan la formación de aminoacil-tRNA), estos anticuerpos tienen una alta especificidad para el complejo polimiositis/dermatomiositis, el más frecuente es el anti -Jo-1 que se observa en un 20% de pacientes de polimiositis, en un 60-70% de pacientes que tienen además fibrosis pulmonar intersticial y en un pequeño porcentaje (5%) de pacientes con dermatomiositis. La presencia de estos anticuerpos define un síndrome "síndrome antisintetasa", caracterizado por la presencia de enfermedad pulmonar intersticial, disnea, manos de mecánico, artritis, fiebre, fenómeno de Raynaud, síndrome del túnel carpiano, polimiositis y dermatomiositis. Los anticuerpos anti-sintetasa casi nunca se observan en pacientes con dermatomiositis asociada a neoplasia ni en dermatomiositis juvenil. Dentro de los anticuerpos musculo-específicos, se ha demostrado que la presencia de anticuerpos contra las proteçinas de 155/140 KD se han relacionado con una alta incidencia de cancer y tambíen se han demostrado en los pacientes con dermatomiositis juvenil. Existe un grupo de anticuerpos que se han detectado en enfermos con síndrome de solapamiento entre miositis-esclerodermia que incluyen los anticuerpos anti PM-SCL, anti -Ku y anti RNP.
Subgrupos de Dermatomiositis
Dermatomiositis juvenil La dermatomiositis juvenil es una forma de miositis inflamatoria acompañada de lesiones cutáneas que se observa en la población infantil y juvenil (2-15 años). Las lesiones cutáneas así como la afectación muscular son de intensidad variable y semejantes a las de la DMS/PM de la población adulta, pero en general tiene un mejor pronóstico. Las lesiones cutáneas más frecuentes son el desarrollo del rash heliotropo periorbitario y en extremidades, eritema periungueal, lesiones psoriasiformes en cuero cabelludo. Este grupo de pacientes desarrolla calcificaciones y vasculitis con mayor frecuencia que la población adulta. Las calcificaciones se observan en un 30-70% de los casos y se observan en codos, rodillas nalgas y zonas traumatizadas pudiendo afectar al tejido subcutáneo, muscular, fascia y exosquelético. Las vasculitis pueden afectar a piel, músculo y tubo digestivo donde se expresan clínicamente en forma de abdomen agudo.
Dermatomiositis amiopátíca: describe un grupo de pacientes con lesiones cutáneas características de dermatomiositis que tras un período de observación no presentan manifestaciones musculares, clínicas, histológicas o con técnicas de imagen (MRI). Representa alrededor del 10% de pacientes con dermatomiositis, afecta predominantemente a adultos jóvenes. La lesión cutánea más característica es el desarrollo de rash heliotropo periorbitario, los pacientes también suelen tener lesiones cutáneas acrales con pápulas y signo de Gottron así como telangiectasias periungueales. Es frecuente el hallazgo de fotosensibilidad marcada, artralgias y fenómeno de Raynaud. En este grupo de pacientes suele referir manifestaciones sistémicas en forma de mal estado general y cansancio, pero los estudios clínicos descartan la afectación muscular.
Dermatomiositis asociada a enfermedades del tejido conectivo: Alrededor de un 20% de pacientes con DM/PM presentan signos de otras enfermedades del tejido conectivo tales como LES, esclerodermia, S. Sjogren, AR, o enfermedad mixta del tejido conectivo, presentando manifestaciones comunes a ambas entidades. Estos pacientes suelen presentar una miositis leve. Serologicamente tienen una mayor tendencia a presentar anticuerpos asociados a la dermatomiositis, pero no especificos, con unos ANA de más de 1:640, anti DNAds, anti ENA( especialmente anti RNP). Los pacientes que presentan el anticuerpo anti PM/SCL engloban un grupo de pacientes que presentan signos clínicos de esclerodermia y dermatomiositis que se han descrito bajo el término de escleromiositis. Son niños que refieren miositis y mialgias, con presencia de dedos escleróticos y fenómenos de Raynaud.
Dermatomiositis y neoplasia maligna: Diversos estudios retrospectivos han descrito la asociación entre dermatomiositis y cáncer con una frecuencia que oscila entre un 10 y un 50%, las neoplasias se observan en asociación con la dermatomiositis del adulto pero no el la forma juvenil ni en la polimiositis. Ciertos tipos de neoplasia como el carcinoma de ovario en las mujeres y gástrico o linfomas en los varones aparecen más frecuentemente en pacientes con dermatomiositis que en la población normal. En pacientes de más de 45 años con DMS deben realizarse estudios clínicos, analíticos y radiológicos para descartar la presencia de una neoplasia.
Tratamiento de la dermatomiositis: La administración sistémica de corticoides es el tratamiento inicial de la dermatomiositis, si la afectación muscular no es muy importante, la administración de dosis bajas (<0,5 mg/kg/d) se relaciona con menos efectos secundarios. En pacientes en que no se controla la enfermedad deben asociarse otros inmunosupresores como el metotrexate y azatioprina, ciclosporina o inmunoglobulinas endovenosas.
http://www.uv.es/derma/CLindex/CLLE/CLlupus.html
Dermatomiositis
El año pasado, la TMA trabajo con la Asociación Americana de Médicos Familiares para llegar a una explicación de miositis que pudiera ser entendida a nivel de 6° año de primaria. Con algunos pequeños cambios, hemos modificado la parte de Dermatomiositis para discutir la Dermatomiositis Juvenil (JM).
Que es la Dermatomiositis ?
La Dermatomiositis (DM) es una enfermedad del músculo. Esta enfermedad puede causar dolor y puede limitar las actividades de una persona debido a la debilidad muscular.
Quien padece DM o PM ?
La DM puede aparecer a cualquier edad pero es más común entre los 40 y 60 años de edad. Una forma de DM ocurre en niños y adolescentes y se conoce como Dermatomiositis Juvenil. La mayoría de los niños con Dermatomiositis Juvenil pueden recuperarse después de uno o más tratamientos.
Que causa la DM?
Se cree que la DM es una enfermedad autoinmune y que es causada por una respuesta inmunológica. El sistema inmunológico del organismo generalmente ataca las infecciones. Cuando el sistema inmune ataca los propios tejidos del organismo, se le llama “enfermedad autoinmune”. Por ejemplo, cuando el sistema inmune ataca los músculos, estos se debilitan. La DM no se propaga de persona a persona y tampoco son enfermedades que se transmitan por herencia directa.
Cuales son los signos de DM ?
La DM causa debilidad, dolor y cansancio. Uno de los signos más importantes de la DM es el eritema cutáneo, que por lo general se presenta antes de tener debilidad muscular. El eritema es de color rojo o violeta y puede ser duro o suave. Puede aparecer en cualquier parte del cuerpo, pero es más común en la cara, los párpados, el cuello, la parte anterior del tórax, nudillos, rodillas y codos. Hay sin embargo personas que tienen eritema cutáneo y no presentan debilidad muscular en años. Algunas personas pueden tener los ojos hinchados.
Como se diagnostica la DM ?
Su medico puede hacer pruebas de reflejos y de tono muscular y examinar el patrón de debilidad muscular. Una prueba de sangre también puede ser de gran utilidad para buscar proteínas de los músculos. Su medico puede también hacer pruebas para determinar la contractilidad muscular. Algunas veces con la biopsia de músculo es suficiente. El diagnosticar la DM en personas que tienen el típico eritema por lo general es fácil. Un diagnostico preciso en personas que no tienen
característicos bien definidos puede resultar una experiencia difícil para el medico y para el paciente.
Como se trata la DM ?
La primera opción de tratamiento para la DM es casi siempre prednisona por vía oral. Este es un medicamento esteroideo que suprime el sistema inmune. Por lo general, se recomienda iniciar tomando una dosis alta durante uno o dos meses. Después, la dosis puede disminuirse durante los siguientes meses.
Puede ser necesario seguir tomando la prednisona durante un año o mas para prevenir que los síntomas vuelvan a aparecer. Su medico decidirá la mejor manera de llevar acabo el tratamiento. Aproximadamente la mitad de las personas con DM o PM no responden adecuadamente a la prednisona y necesitaran tomar otros medicamentos.
Como se trata el eritema cutáneo de la DM ?
El eritema por lo general mejora con el tratamiento medico. Si éste no desaparece, su medico podría recetarle hidroxicloroquina (nombre comercial: Plaquenil). Este medicamento también causa supresión del sistema inmune. Su medico también puede recetarle un corticosteroide. Debido a que el eritema puede empeorar con la exposición a la luz solar, deberá de evitarse la exposición directa al sol y utilizar bloqueadores solares cuando sea necesario.
Como se puede tratar el dolor ?
Los tratamientos más comunes para el dolor son medicamentos como la aspirina, ibuprofeno (nombre comercial: Nuprin), acetaminofeno (Tylenol) y naproxeno (Aleve). Si esto no alivia el dolor, su medico puede prescribirle una medicina mas potente. Otros tratamientos que pueden ser útiles incluyen baños calientes, la aplicación de bolsas calientes o frías, y ejercicios de estiramiento.
Como puede tener una mejor vida una persona con DM ?
Su medico puede preguntarle a un terapeuta físico que le enseñe a hacer ejercicios de estiramiento para mejorar su movilidad. Estos ejercicios pueden ayudar a mantener la movilidad en los músculos que estén más débiles. El uso de bastón, andadera o silla de ruedas puede ayudar a las personas que tienen problemas para movilizarse. También se pueden usar en casos más graves aparatos especiales para las piernas, cuello o muñecas.
Asociación Americana de Miositis
1233 20th Street NW, Suite 402
Washington DC 20036
Teléfono: (202) 887-0088
Telefax: (202) 466-8940
Dirección de correo electrónico: [email protected]
Pagina en Internet: http://www.myositis.org
http://afibro.org/2011/07/dermatomiositis/
http://www.blogmedicina.com/2008/08/10/dermatomiositis-juvenil/
http://www.ferato.com/wiki/index.php/Dermatomiositis
http://lanoticiaperu.blogspot.com.es/2009_01_01_archive.html
RESUMEN
La Dermatomiositis amiopática es una variante de dermatomiositis en la cual los pacientes tienen manifestaciones cutáneas sin evidencia clínica de afectación muscular y en los que las pruebas complementarias no demuestran ningún grado de inflamación muscular durante más de 2 años. Se presenta un paciente varón de 47 años de edad que acude al consultorio de dermatología con lesiones eritematosas en cara, brazos, pecho y espalda, levemente pruriginosas, no dolorosas, fotosensibilidad, cambios en la piel de articulaciones de las manos y codos, acompañados de caída de cabello de mas de dos años de evolución sin compromiso neurológico motor. Los exámenes de laboratorio confirman el diagnóstico de dermatomiositis amiopática.
Palabras clave: Dermatomiositis amiopatica, colagenopatias, polimiositis, autoinmunidad, corticoterapia sistemica
INTRODUCCIÓN
La dermatomiositis (DM) es un desorden multisistémico de etiología desconocida, caracterizada clásicamente por miopatía inflamatoria idiopática asociada a manifestaciones cutáneas características.
En 1975, Bohan y Meter presentan una serie de criterios que permitirían establecer el diagnóstico y clasificación de dermatomiositis. Cuatro de ellos aluden al compromiso muscular: debilidad simétrica progresiva y proximal, aumento de la concentración de enzimas musculares, electromiografía anormal y una biopsia de tejido muscular anormal. El quinto punto hace referencia al compromiso cutáneo(1).
La clasificación clínica de la dermatomiositis empleada en la actualidad(2) es la siguiente
Grupo I: Polimiositis idiopática primaria: puede aparecer a cualquier edad. Se manifiesta exclusivamente con síntomas musculares.
Grupo II: Dermatomiositis idiopática primaria: asocia sintomatología cutánea y muscular.
Grupo III: Dermatomiositis o polimiositis asociada a neoplasia: las alteraciones clínicas son indistinguibles de las de la DM de cualquier otro tipo. La incidencia del síndrome es mayor en pacientes con edades superiores a los 60 años.
Grupo IV: Dermatomiositis o polimiositis infantil: presenta una mortalidad elevada (en torno al 33%), aunque en general el pronóstico suele ser mejor que el de las formas del adulto.
Grupo V: Dermatomiositis o polimiositis asociada a conectivopatías: las enfermedades asociadas con más frecuencia son la esclerosis sistémica progresiva, la artritis reumatoide, el lupus eritematoso y la conectivopatía mixta.
La severidad del compromiso muscular y sus secuelas, la afección visceral, la asociación con neoplasias y la respuesta a corticosteroides, son los principales factores pronósticos de este grupo de entidades(3). La debilidad muscular fue la manifestación inicial en 79% de los pacientes, en la mitad de ellos asociada a lesiones cutáneas características de la DM(3).
La dermatomiositis amiopática ha sido una entidad bastante controvertida en la literatura mundial, porque un porcentaje significativo de los pacientes presentan la miopatía durante el seguimiento(3). La DM es una enfermedad necrosante del músculo estriado asociada con lesiones inflamatorias en la piel. La dermatomiositis amiopática representa un subgrupo de DM en el cual las manifestaciones cutáneas clásicas se presentan durante períodos prolongados en ausencia de enfermedad muscular(3).
Esta particularidad suele conducir a un retraso en el diagnóstico y a confusión en el manejo terapéutico, en que, como sucede en la DM clásica, es de crucial importancia la búsqueda de la patología maligna asociada(4).
***
La dermatomiositis amiopática o dermatomiositis sine miositis es un tipo de dermatomiositis caracterizada por lesiones cutáneas específicas de DM, confirmadas histológicamente, sin evidencia clínica de debilidad muscular proximal, con enzimas musculares normales, de más de seis meses de evolución(10,11). Algunos pacientes tienen hallazgos anormales en la ecografía, RMN o la biopsia muscular. Estos pacientes tienen alteración muscular subclínica, pero su condición puede ser clasificada como dermatomiosoitis amiopática(10,11). En nuestro caso tenemos un paciente que cursa con afectación cutánea, con exámenes de laboratorio y electromiografía sugerente de afectación muscular subclínica. Lo que algunos autores llaman dermatomiositis hipomiopática, para otros es catalogada de premiopática, como lo veremos más adelante. Los criterios de exclusión de dermatomiositis amiopática son: 1) tratamiento con terapia inmunosupresora sistémica por dos meses consecutivos o más, dentro de los primeros seis meses de enfermedad y 2) el uso de fármacos capaces de producir cambios cutáneos similares a la dermatomiositis, como la hidroxiurea y las estatinas hipocolesterelomiantes al comienzo de la afección(10,11).
La dermatomiositis hipomiopática(12) es un tipo de DM con afectación muscular subclínica, en los que las pruebas complementarias como las enzimas musculares, el electromiograma, la resonancia magnética o la biopsia muscular demuestran algún grado de inflamación muscular. Con fines prácticos de manejo terapéutico se agrupan la dermatomiositis amiopática e hipomiopática en la dermatomiositis amiopática clásica(9).
La dermatomiositis amiopática o hipomiopática está confirmada cuando su evolución es mayor de dos años y es provisional cuando la duración es menor de dos años pero mayor de seis meses(12). Este límite de tiempo se propone arbitrariamente tras la observación de que en la DM clásica las manifestaciones cutáneas suelen preceder en semanas o meses a la debilidad muscular (50-60%) y es raro que esta última se desarrolle después de los dos años(11). En nuestro paciente por el tiempo de evolución mayor de dos años de las lesiones cutáneas sin debilidad muscular clínicamente evidente, se considera un diagnostico de dermatomiositis amiopática confirmada, el hallazgo de alteraciones enzimáticas y en la electromiografía hace que no sea necesaria la realización de la biopsia muscular para confirmar el diagnóstico.
Manifestaciones Cutáneas
Aparecen sólo en la DM y acompañan o, más frecuentemente, preceden a las manifestaciones musculares, ocasionalmente hasta en varios años. Existen dos lesiones patognomónicas de dermatomiositis: 1) el eritema en heliotropo, que consiste en una coloración violácea de párpados superiores, asociada muchas veces a edema; lo presentan menos del 50% de los pacientes; y 2) las pápulas de Gottron, unas lesiones pápulo eritematosas descamativas sobre la piel de las articulaciones metacarpofalángicas e interfalángicas, que aparecen en el 60- 80% de los casos. También se observan frecuente-mente otro tipo de lesiones cutáneas que no son específicas, como las erupciones en áreas fotoexpuestas (cuero cabelludo, cara, cuello, espalda, tórax anterior, hombros) que, a diferencia del lupus eritematoso sistémico, pueden ser intensamente pruriginosas. A excepción del eritema en heliotropo estas lesiones fueron evidentes en el caso que presentamos. Son también comunes el eritema y las anomalías capilares periungueales (de forma similar a la esclerodermia), la hipertrofia cuticular o las fisuras palmares o digitales (“manos de mecánico”). Especialmente en las formas juveniles de DM pueden desarrollarse calcificaciones subcutáneas(13).
Exámenes de Laboratorio
En las miopatías inflamatorias (MI), se suelen detectar concentraciones séricas elevadas de creatincinasa (CK), aldolasa, transaminasas (AST, ALT) y lactato deshidrogenasa (LDH), pero sólo la CK es específica para el músculo. Sin embargo, un 10-20% de pacientes tienen enzimas musculares normales. La aldolasa puede elevarse entrastornos hematológicos y hepáticos. Estos últimos también provocan elevaciones de las transaminasas. La hipoxia, los traumatismos musculares (incluyendo inyecciones intramusculares o electromigrafías), el ejercicio vigoroso o ciertos fármacos aumentan las concentraciones de CK. Una elevación aislada de CK en un paciente que no tiene debilidad muscular ni lesiones cutáneas no requiere, por lo general, una evaluación extensa(10).
Anticuerpos antinucleares
Los anticuerpos antinucleares (ANA) son un grupo heterogéneo de autoanticuerpos con reactividad predominante contra antígenos nucleares, pero también contra otros localizados en el citoplasma(10,14).
Los ANA se determinan habitualmente por una técnica de inmunofluorescencia indirecta (FANA) utilizando como substrato células Hep-2 (células de carcinoma laríngeo humano). La positividad se traduce en la observación de una fluorescencia verdosa en el núcleo de las células. Es importante consignar el recíproco de la máxima dilución del suero en la que se observa dicha fluorescencia (título) y el patrón morfológico de la misma(10,14).
Se considera que los ANA son positivos si el título es =1/160. Se debe tener en cuenta que estos autoanticuerpos están presentes no sólo en enfermedades autoinmunes sistémicas, sino también en procesos autoinmunes órgano específicos, infecciones, neoplasias e incluso en la población normal; los pacientes con dermatomiositis amiopática tienen niveles anormalmente elevados de anticuerpos antinucleares con un patrón de fluorescencia nuclear moteado(4).
En un estudio realizado por Sontheimer y Targoff(9) se observó que 16 de 18 muestras de suero (89%) de pacientes con dermatomiositis amiopática contenían anticuerpos recientemente identificados por inmunoblot e inmunoprecipitación contra una proteína de 155 kDa y/o la proteína Se. Ninguno de ellos presentó anticuerpos específicos de miositis (anti-Jo 1 y Mi 2). Si se confirman estos hallazgos, los anticuerpos específicos anti-p155 y anti-Se podrían identificar a un subgrupo de pacientes con DM que tienden a permanecer amiopáticos(4).
La Biopsia de Piel y de Músculo
Los cambios histológicos en piel son bastante variables, Unas veces los cambios son sutiles y solo se aprecia un infiltrado perivascular superficial parcheado de linfocitos, asociado a edema variable y cambios mucinosos en la dermis superficial, como se observa en nuestro paciente. Más frecuentemente existen las características de un patrón de reacción tisular que consta de cambios vacuolares en la capa basal, presentando rasgos indistinguibles del lupus eritematoso agudo(15).
La biopsia de músculo no solo sirve para confirmar la presencia de una afectación muscular y establecer el diagnóstico de polimiositis, DM o miositis a cuerpos de inclusión, sino también para excluir otras enfermedades neuromusculares. Los elementos que permiten el diagnóstico de una miopatía inflamatoria en la microscopía óptica son: 1) infiltrados de células inflamatorias en el tejido muscular: 2) fibras musculares en degeneración, necrosis, fagocitosis y regeneración: 3) Incremento del tejido conjuntivo endo y perimisial. La presencia, localización y distribución de los hallazgos son distintos para cada una de las entidades consideradas(16). Nuestro paciente no permitió la realización de este examen.
La Electromiografía
En las miopatías inflamatorias idiopáticas, el electromiograma será compatible con un proceso miopático activo, aunque en la miopatía por cuerpos de inclusión también se observan datos de neuropatía concomitante. No obstante, el electromiograma es normal en el 15% de los casos de Miopatías inflamatorias idiopáticas. El electromiograma puede sugerir cuál es el músculo apropiado que debe biopsiarse como también la resonancia magnética. Dado que esta prueba puede inducir áreas locales de inflamación se debe realizar en un lado del cuerpo, reservando el otro hemicuerpo para la biopsia(10). Esta prueba arrojó cambios leves sugestivos de miopatía en nuestro caso.
La Resonancia Magnética de Partes Blandas
La resonancia magnética de partes blandas de las cinturas escapular y pelviana permite identificar inflamación muscular subclínica al detectar hiperintensidad en la secuencia T2 y sería más sensible (97%) que la biopsia muscular, aunque menos específica. La resonancia magnética es útil además como guía de una eventual biopsia por la ya referida distribución en parches y en el seguimiento. Si la resonancia magnética exhibiera cambios, se requeriría ampliar el estudio con un electromiograma y/o biopsia muscular. Se describe también el uso de la ecografía, el centellograma con talio y la espectroscopia con fósforo(4).
Tratamiento
Los corticosteroides son los medicamentos de primera elección y continúan siendo la base del tratamiento. La dosis a las cuales los usamos son dosis divididas de prednisona equivalentes a 60 mg/día, en los pacientes con dermatomiositis/polimiositis. El metotrexate y los medicamentos antipalúdicos son ahorradores de corticoides y se pueden usar desde el inicio de modo conjunto, luego se pueden ir retirando gradualmente los corticoides, tal como se hizo con este paciente(10,11).
En pacientes con dermatomiositis, con miositis definida, se comprobó la importancia del tratamiento precoz con corticoides sistémicos, pero éstos no siempre son eficaces para tratar las lesiones cutáneas. Existe consenso en no tratar en un comienzo a los pacientes con dermatomiositis amiopática con terapias sistémicas agresivas, y sí con fotoprotección, corticoides tópicos y antipalúdicos (hidroxicloroquina 200-400 mg/día o cloroquina 250 mg/día), bajo control oftalmológico, hematológico y de la función hepática. Una alternativa terapéutica en los pacientes que no responden a este esquema es el metotrexate en dosis bajas (2,5-30 mg/ semana), mientras que los corticoides sistémicos se reservan para casos refractarios(12,13). Últimamente se han descrito casos de dermatomiositis refractarios a prednisona y antimaláricos, que han respondido bien a terapia con dapsona, constituyéndose en una terapia alternativa importante por su bajo costo y fácil accesibilidad( 17). Si existe prurito, es posible agregar antihistamínicos. Con respecto a los autoanticuerpos, los que tienen los anti SRP son los de peor pronóstico, los antisintetasas se asocian con recurrencias y disminución de la sobrevida. Los pacientes con anti Mi-2 tienen una sobrevida a cinco años que alcanza el 95%(10,14).
http://revistas.concytec.gob.pe/scielo.php?pid=S1028-71752008000100008&script=sci_arttext
Manifestaciones clínicas y diagnóstico de la dermatomiositis (DM) y la polimiositis (PM) en adultos.
Introducción:
La dermatomiositis (DM) y la polimiositis (PM) son clasificadas como miopatías inflamatorias (1-3) La prevalencia de ambos trastornos es de 1/100.000 en la población general. Hay una predilección por el sexo femenino en una relación de 2/1. El pico de incidencia en adultos ocurre entre los 40 y los 50 años, pero puede comenzar a cualquier edad. (4-5)>
Diferencias entre la DM y la PM
Aunque la DM y la PM comparten la debilidad muscular, hay diferencias tanto clínicas como patogénicas entre ambos trastornos:
La DM se asocia a una variedad de manifestaciones en piel, incluyendo el signo de Gottron (piel eritematosa, engrosada y descamativa en los nudillos), el signo del manto o del chal (lesiones eritematosas en la “V” del cuello y en la parte anterior y posterior del tórax así como los hombros, justamente en la zonas
que cubre un chal), el exantema en heliotropo (coloración lila o violácea en los párpados), y la eritrodermia generalizada.
La DM se asocia más probablemente a cáncer que la PM.
La DM se caracteriza por deposición de inmunocomplejos en los vasos y es considerada en parte, como una vasculopatía mediada por complemento. En contraste, la PM parece reflejar la injuria muscular directa, mediadas por células-T.
Criterios de clasificación:
>Varios sets de clasificación han sido desarrollados para clasificar la DM y la PM.
Criterios de Bohan y Peter
Los criterios originales de Bohan y Peter formulados en 1975 incluyen los siguientes elementos:
1) Debilidad muscular proximal simétrica.
2) Rash típico de DM, que es el único hallazgo distintivo entre la DM y la PM.
3) Enzimas musculares elevadas.
4) Cambios miopáticos en la electromiografía.
5) Hallazgos característicos en la biopsia muscular, y ausencia de signos histopatológicos de otras miopatías.
http://www.elrincondelamedicinainterna.com/2009/08/dermatomiositis-en-una-mujer-de-47-anos.html
DEFINICIÓN
Enfermedad de etiología autoinmunitaria que
afecta fundamentalmente al músculo esquelético y
a la piel. Se caracteriza por un proceso inflamatorio
no supurativo con predominio de inflamación
linfocitaria. Se denomina polimiositis cuando se
respeta la piel y dermatomiositis cuando la polimiositis
se asocia a una erupción cutánea característica.
La tercera parte de los casos se asocian a
diferentes enfermedades del tejido conectivo y una
décima parte a neoplasias malignas.
ETIOPATOGENIA
La causa es desconocida, pero parece que
contribuyen varios factores: 1. Factores genéticos.
Una mayor predisposición genética para
HLA DR3 y DRW52. 2. Mecanismo inmunitario.
La presencia de autoanticuerpos circulantes frente
antígenos musculares (anti-JO 1, anti-Mi, anti-
PM1, anti-PM/Scl), linfocitos CD8 y macrófagos
que invaden fibras musculares. 3. Virus. Se han
involucrado distintas partículas víricas en su
etiología. Es más frecuente en mujeres (excepto
el tipo III) y en edades comprendidas entre los
45 y 60 años.
http://www.e-dermatosis.com/pdf-zip/Derma026.pdf
La dermatomiositis es una enfermedad del tejido conectivo, caracterizado por inflamación de los músculos y de la piel. Su causa es desconocida, pero puede resultar de una infección viral o una reacción autoinmune. Más del 50% de los casos puede ser un fenómeno paraneoplástico, indicando presencia de cáncer. Se identifica por un rash característico, acompañado de debilidad muscular.
Los rayos X ilustran calcificaciones distróficas en los músculos. Hay una forma de este desorden que ataca a los niños, conocida como dermatomiositis juvenil. Las pápulas de Gottron son parches rosados en los nudillos, asociados con este desorden.
http://es.wikipedia.org/wiki/Dermatomiositis
http://www.nlm.nih.gov/medlineplus/spanish/ency/article/000839.htm
http://www.revistacolombianadereumatologia.org/Portals/0/Descargas/9d-dermatomiositis 10-2.pdf
http://www.cnoo.es/modulos/gaceta/actual/gaceta390/cientifico%201.pdf
http://www.pathologyoutlines.com/topic/skinnontumordermatomyositis.html
Fármacos indicados:
Vídeo:
Visitas: 17890
Galería:







Mostrando Registros desde el 1 hasta el 0 de un total de 0















 Boletín
Boletín

|

|
Últimos Comentarios Nanci Gonzalez  Eflornitina Quiero esta crema... alvaro gil  Triamcinolona donde consigo este medicamento Nombre:... Hugo  Griseofulvina Me interesa comprarlo.. Donde puedo adquirirlo?... daniels  Ácido Salicílico / Ácido Láctico me pueden ayudar me cayo en la cara y ciertas partes se volvieron moradas pueden decirme como se... Chiquitaberry  Dermatitis crónica de manos y pies Por favor ayúdenme soy de Venezuela y no hay medicamento que tratamiento debo usar para curar las... |
Dermas más visitados |
Tratamientos más visitados Ácido Salicílico / Ácido Láctico  160278 Hidroquinona  151607 Cloruro de Aluminio  77346 Ácido fusídico  67294 Miconazol  58739 |

|




